Programs in the Lab
The Thayer Labratory uses many different programs to assist their research. Below are highlighted publications that actively use programs commonly utilized by the Thayer Labratory.
Publications that Utilize Thayer Labratory Programs
-
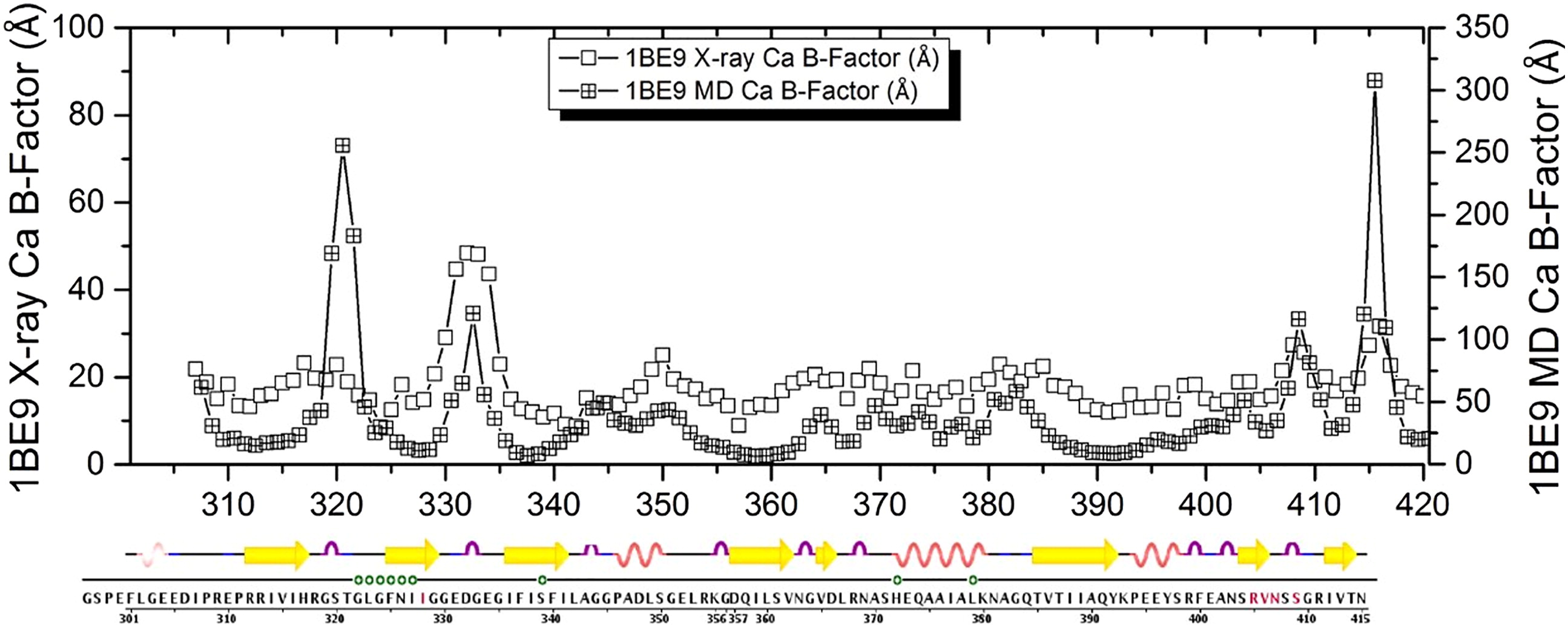 Spectral analysis of Molecular Dynamics computer simulations on the PDZ protein: MD: Sectors.
Spectral analysis of Molecular Dynamics computer simulations on the PDZ protein: MD: Sectors.
Lakhani B, Thayer KM, Beveridge DL.
Journal of Biomolecular Structure and Dynamics (2019). DOI: 10.1080/07391102.2019.1588169
The idea of protein “sectors” posits that sparse subsets of amino acid residues form cooperative networks that are key elements of protein stability, ligand binding, and allosterism. To date, protein sectors have been calculated by the statistical coupling analysis (SCA) method of Ranganathan and co-workers via the spectral analysis of conservation-weighted evolutionary covariance matrices obtained from a multiple sequence alignments of homologous families of proteins. SCA sectors, a knowledge-based protocol, have been indentified with functional properties and allosterism for a number of systems. -
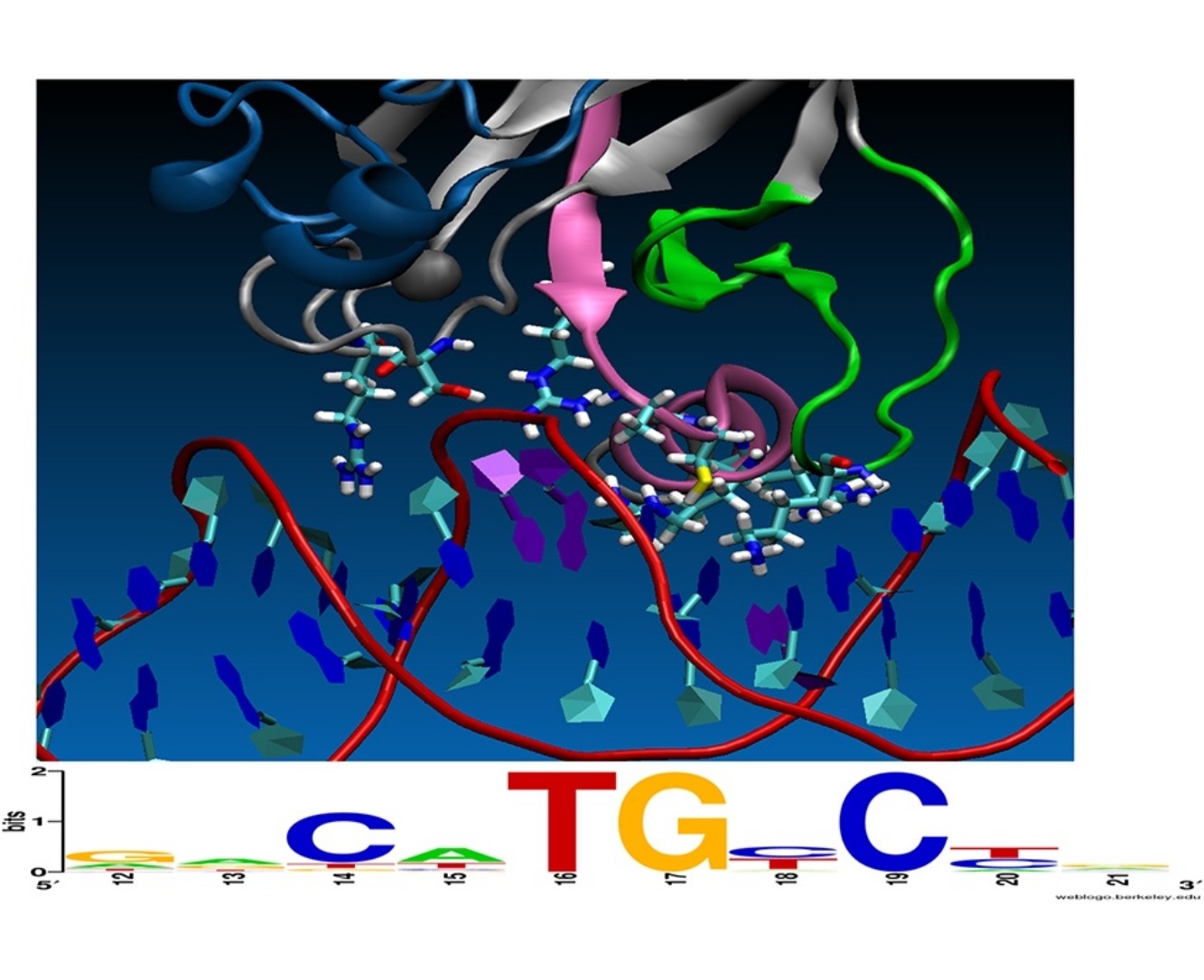 Chemical Principles Additive Model Aligns Low Consensus DNA Targets of p53 Tumor Suppressor Protein.
Chemical Principles Additive Model Aligns Low Consensus DNA Targets of p53 Tumor Suppressor Protein.
Thayer KM and Han IM*.
Computational Biology and Chemistry, 68, 186-193 (2017). DOI 10.1016/j.compbiolchem.2017.03.003
Computational prediction of the interaction between protein transcription factors and their cognate DNA binding sites in genomic promoters constitutes a formidable challenge in two situations: when the number of discriminatory interactions is small compared to the length of the binding site, and when DNA binding sites possess a poorly conserved consensus binding motif. The transcription factor p53 tumor suppressor protein and its target DNA exhibit both of these issues. -
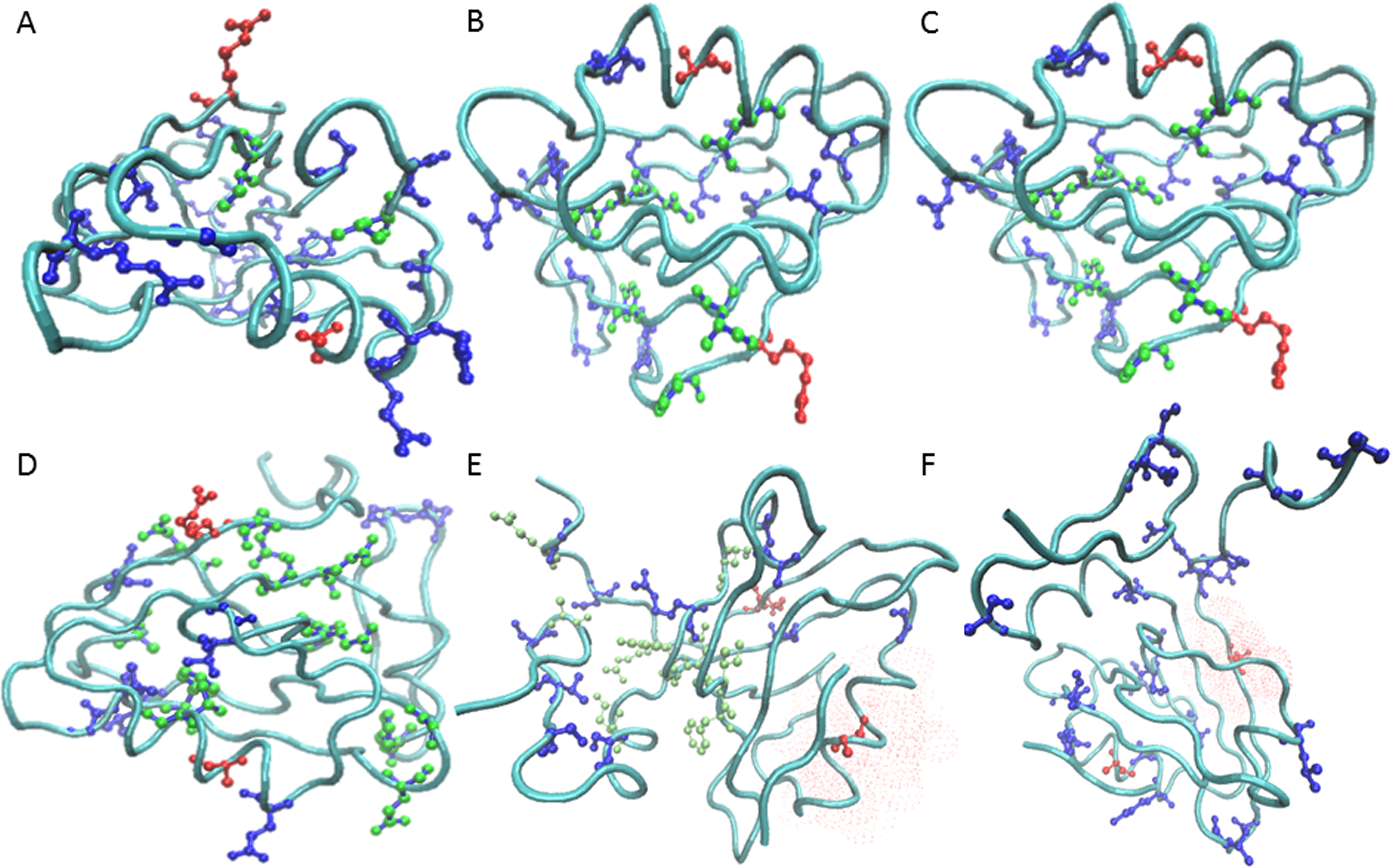 Dependence of prevalence of contiguous pathways in proteins on structural complexity.
Dependence of prevalence of contiguous pathways in proteins on structural complexity.
Thayer KM, Galganov JC*, Stein AJ*.
PLoS ONE, 12, e0188616 (2017). DOI 10.1371/journal.pone.0188616
Allostery is a regulatory mechanism in proteins where an effector molecule binds distal from an active site to modulate its activity. Allosteric signaling may occur via a continuous path of residues linking the active and allosteric sites, which has been suggested by large conformational changes evident in crystal structures. An alternate possibility is that the signal occurs in the realm of ensemble dynamics via an energy landscape change. While the latter was first proposed on theoretical grounds, increasing evidence suggests that such a control mechanism is plausible. -
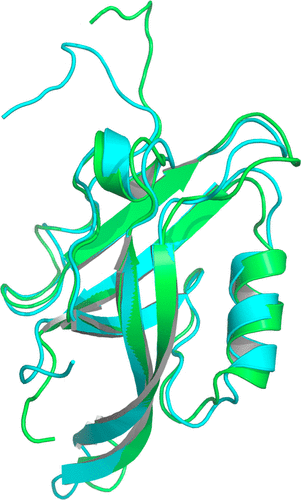 A Molecular Dynamics-Markov State Model of Protein Ligand Binding and Allostery in CRIB-PDZ: Conformational Selection and Induced Fit.
A Molecular Dynamics-Markov State Model of Protein Ligand Binding and Allostery in CRIB-PDZ: Conformational Selection and Induced Fit.
Thayer KM, Lakhani B, and Beveridge DL.
The Journal of Physical Chemistry B.,121, 5509-5514 (2017). DOI: 10.1021/acs.jpcb.7b02083
Conformational selection and induced fit are well-known contributors to ligand binding and allosteric effects in proteins. Molecular dynamics (MD) simulations now enable the theoretical study of protein–ligand binding in terms of ensembles of interconverting microstates and the population shifts characteristic of “dynamical allostery.” Here we investigate protein–ligand binding and allostery based on a Markov state model (MSM) with states and rates obtained from all-atom MD simulations.