Full List of Publications by the Lab
-
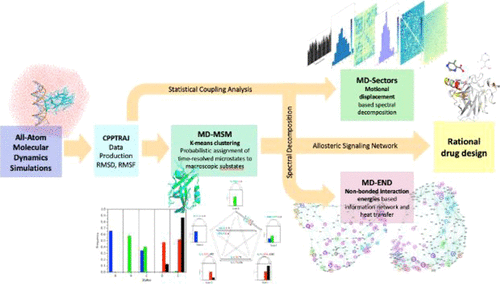 Insights into Rational Design of a New Class of Allosteric Effectors with Molecular Dynamics
Insights into Rational Design of a New Class of Allosteric Effectors with Molecular Dynamics
Han ISM, Abramson D*, Thayer KM.
Markov State Models and Network Theory.
ACS Omega,7(2831) (2022). DOI: 10.1021/acsomega.1c05624.
The development of drugs to restore protein function has been a major advance facilitated by molecular medicine. Allosteric regulation, a phenomenon widely observed in nature, in which a molecule binds to control a distance active site, holds great promise for regulating proteins, yet how to rationally design such a molecule remains a mystery. -
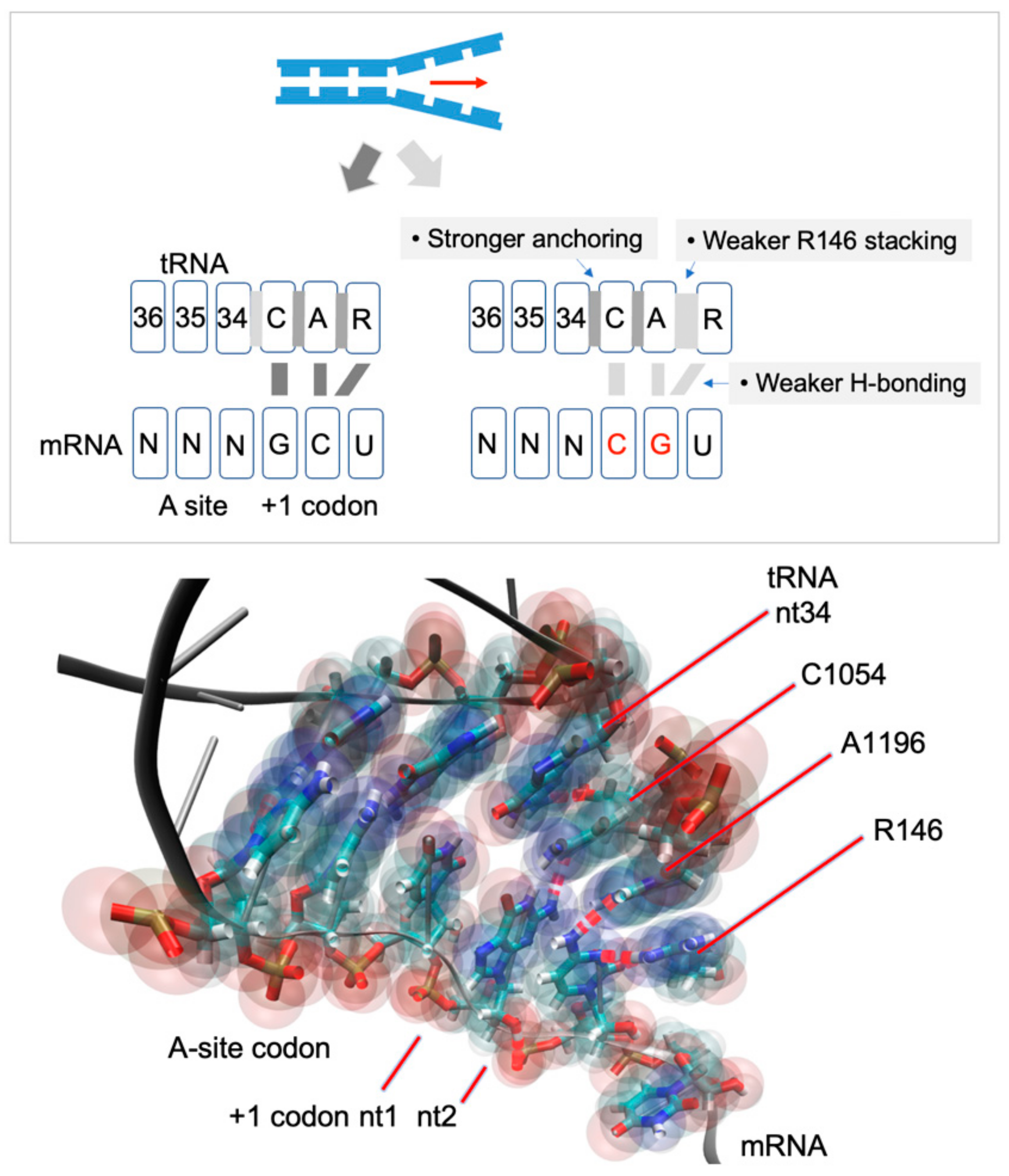 The CAR-mRNA Interaction Surface Is a Zipper Extension of the Ribosome A Site.
The CAR-mRNA Interaction Surface Is a Zipper Extension of the Ribosome A Site.
Dalgarno C*, Scopino K, Raval M, Nachmanoff C, Sakkas ED, Krizanc D, Thayer KM, Weir MP.
Int J Mol Sci, 23(1417) (2022). DOI: 10.3390/ijms23031417.
The ribosome CAR interaction surface behaves as an extension of the decoding center A site and has H-bond interactions with the +1 codon, which is next in line to enter the A site. Through molecular dynamic simulations, we investigated the codon sequence specificity of this CAR–mRNA interaction and discovered a strong preference for GCN codons, suggesting that there may be a sequence-dependent layer of translational regulation dependent on the CAR interaction surface. -
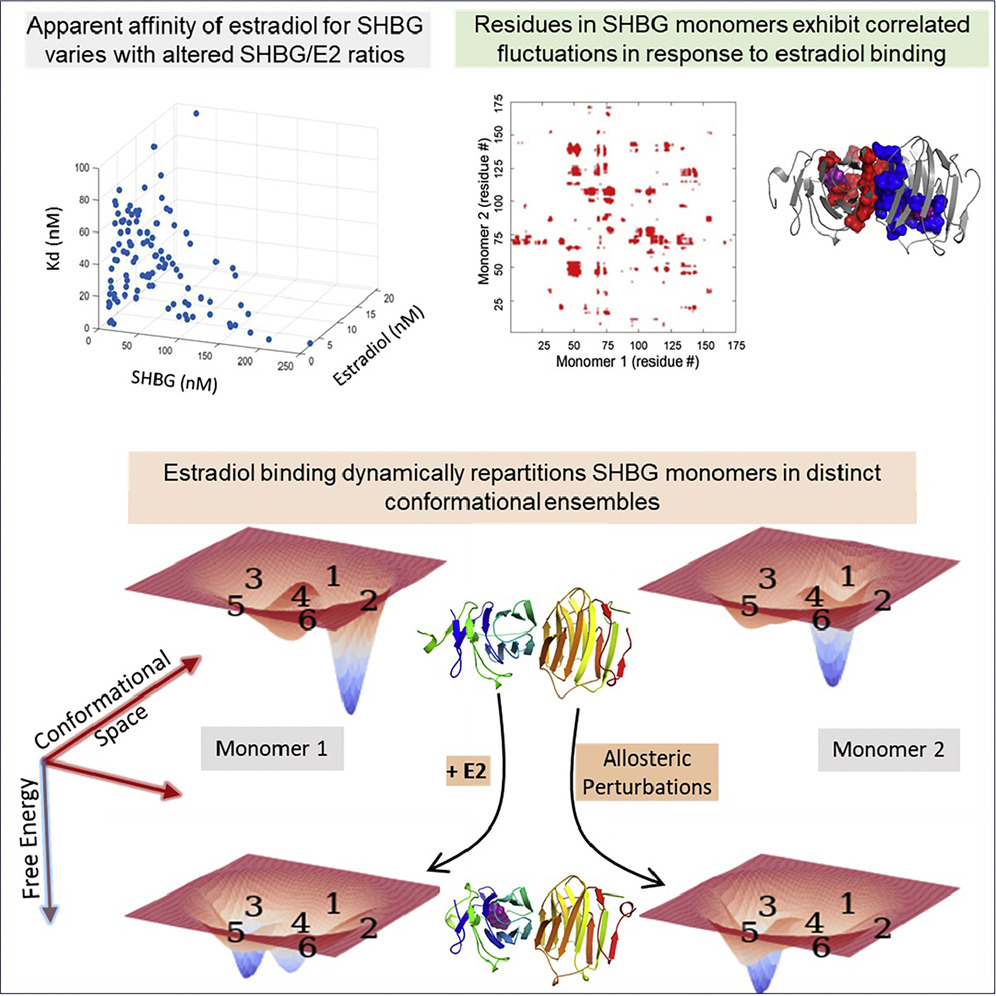 Estradiol Binding Induces Allosteric Coupling and Partitioning of Sex Hormone Binding
Estradiol Binding Induces Allosteric Coupling and Partitioning of Sex Hormone Binding
Jasuja R, Spencer DJ, Jayaraj A, Peng L, Krishna M, Lawney B, Patel P, Jayaram B, Thayer KM,
Beveridge DL, Bhasin S.
Globulin Monomers Among Various Conformational States.
iScience, 24(102424) (2021). DOI: 10.1016/j.isci.2021.102414
Sex-hormone-binding globulin (SHBG) regulates the transport and bioavailability of estradiol. The dynamics of estradiol's binding to SHBG are incompletely understood, although it is believed that estradiol binds to each monomer of SHBG dimer with identical affinity (Kd ∼2 nM). Contrary to the prevalent view, we show that estradiol's binding to SHBG is nonlinear, and the "apparent" Kd changes with varying estradiol and SHBG concentrations. -
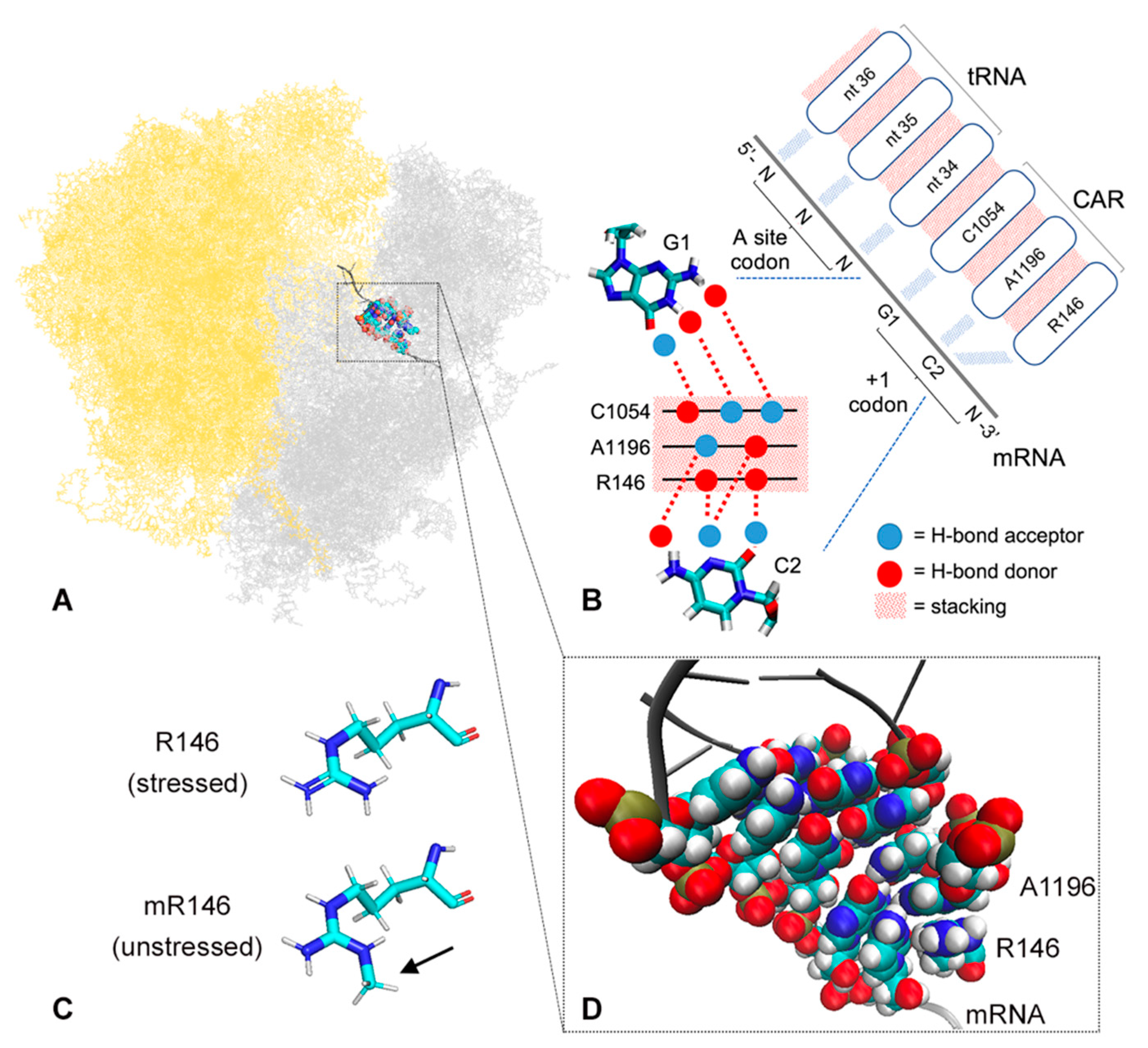 Arginine Methylation Regulates Ribosome CAR Function.
Arginine Methylation Regulates Ribosome CAR Function.
Scopino K*, Dalgarno C*, Nachmanoff C*, Krizanc D, Thayer KM, Weir MP.
International Journal of Molecular Science 22, 1335 (2021). DOI: 10.3390/ijms22031335
The ribosome CAR interaction surface is hypothesized to provide a layer of translation regulation through hydrogen-bonding to the +1 mRNA codon that is next to enter the ribosome A site during translocation. The CAR surface consists of three residues, 16S/18S rRNA C1054, A1196 (E. coli 16S numbering), and R146 of yeast ribosomal protein Rps3. R146 can be methylated by the Sfm1 methyltransferase which is downregulated in stressed cells. -
 GCN sensitive protein translation in yeast.
GCN sensitive protein translation in yeast.
Barr WA*, Sheth RB*, Kwon J*, Cho J, Glickman JW, Hart F, Chatterji OK, Scopino K*, Voelkel-
Meiman K, Krizanc D, Thayer KM, Weir MP.
PLoS One, 15(9) (2020). DOI: 10.1371/journal.pone.0233197. PMCID: PMC7500604
Levels of protein translation by ribosomes are governed both by features of the translation machinery as well as sequence properties of the mRNAs themselves. We focus here on a striking three-nucleotide periodicity, characterized by overrepresentation of GCN codons and underrepresentation of G at the second position of codons, that is observed in Open Reading Frames (ORFs) of mRNAs. Our examination of mRNA sequences in Saccharomyces cerevisiae revealed that this periodicity is particularly pronounced in the initial codons-the ramp region-of ORFs of genes with high protein expression. -
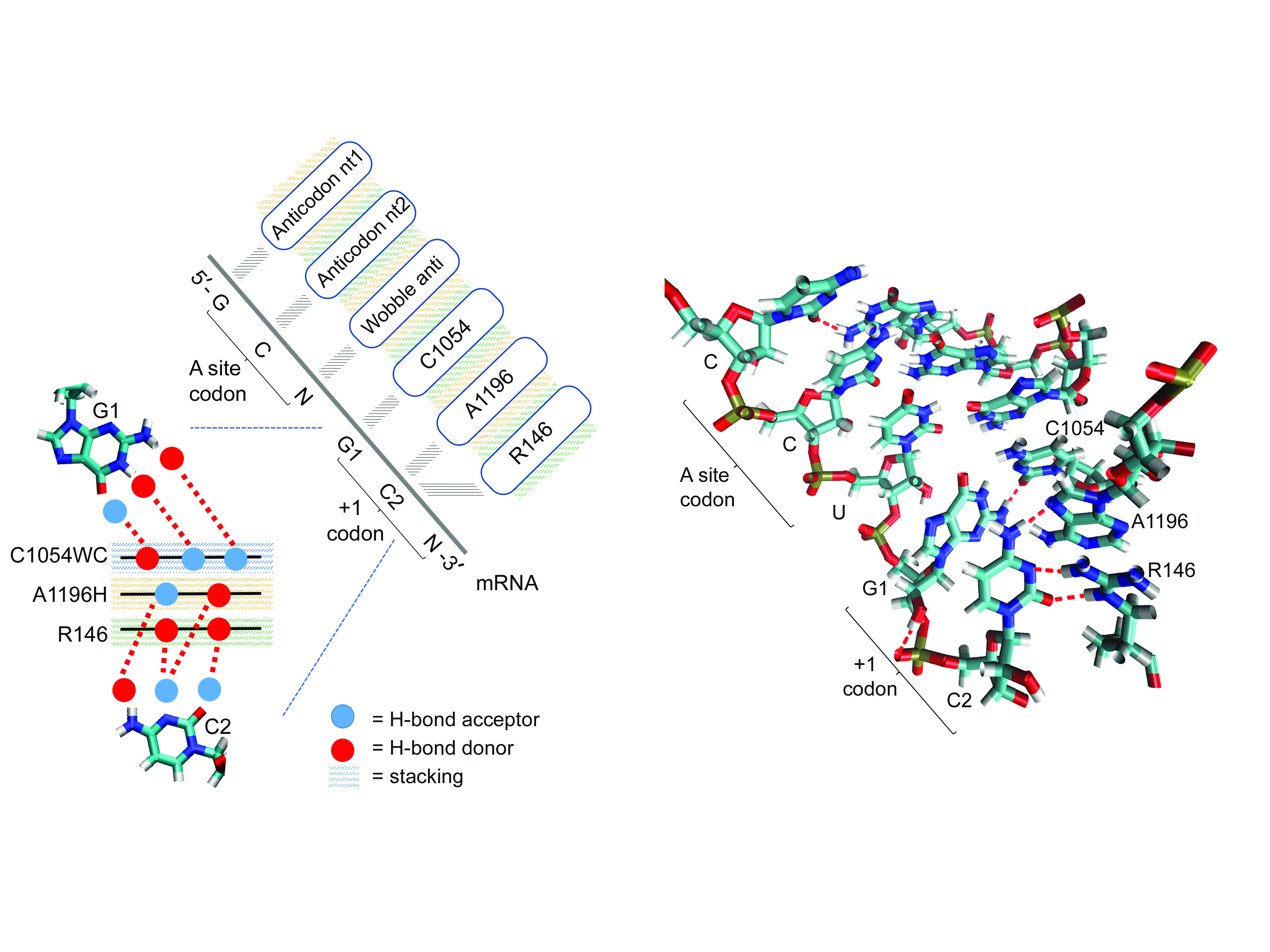 A Ribosome Interaction Surface Sensitive to mRNA GCN Periodicity.
A Ribosome Interaction Surface Sensitive to mRNA GCN Periodicity.
Scopino K*, Williams E*, Elsayed A*, Barr WA*, Krizanc D, Thayer KM, Weir MP.
Biomolecules, 10(6) (2020). DOI: 10.3390/biom10060849
A longstanding challenge is to understand how ribosomes parse mRNA open reading frames (ORFs). Significantly, GCN codons are over-represented in the initial codons of ORFs of prokaryote and eukaryote mRNAs. We describe a ribosome rRNA-protein surface that interacts with an mRNA GCN codon when next in line for the ribosome A-site. The interaction surface is comprised of the edges of two stacked rRNA bases: the Watson–Crick edge of 16S/18S rRNA C1054 and the adjacent Hoogsteen edge of A1196 (Escherichia coli 16S rRNA numbering). -
 Homologs of the Tumor Suppressor Protein p53: A Bioinformatics Study for Drug Design
Homologs of the Tumor Suppressor Protein p53: A Bioinformatics Study for Drug Design
Thayer KM, Carcamo C*.
MOJ Proteomics and Bioinformatics, 9, 5-14 (2020). DOI: 10.15406/mojpb.2020.09.0027
Sequence and structure of proteins related to the tumor suppressor protein p53 were studied from the perspective of gaining insight for the development of therapeutic drugs. Our study addresses two major issues encumber bringing novel drugs to market: side effects and artifacts from animal models. In the first phase of our study, we performed a genome-wide search to identify potentially similar proteins to p53 that may be susceptible to off target effects. -
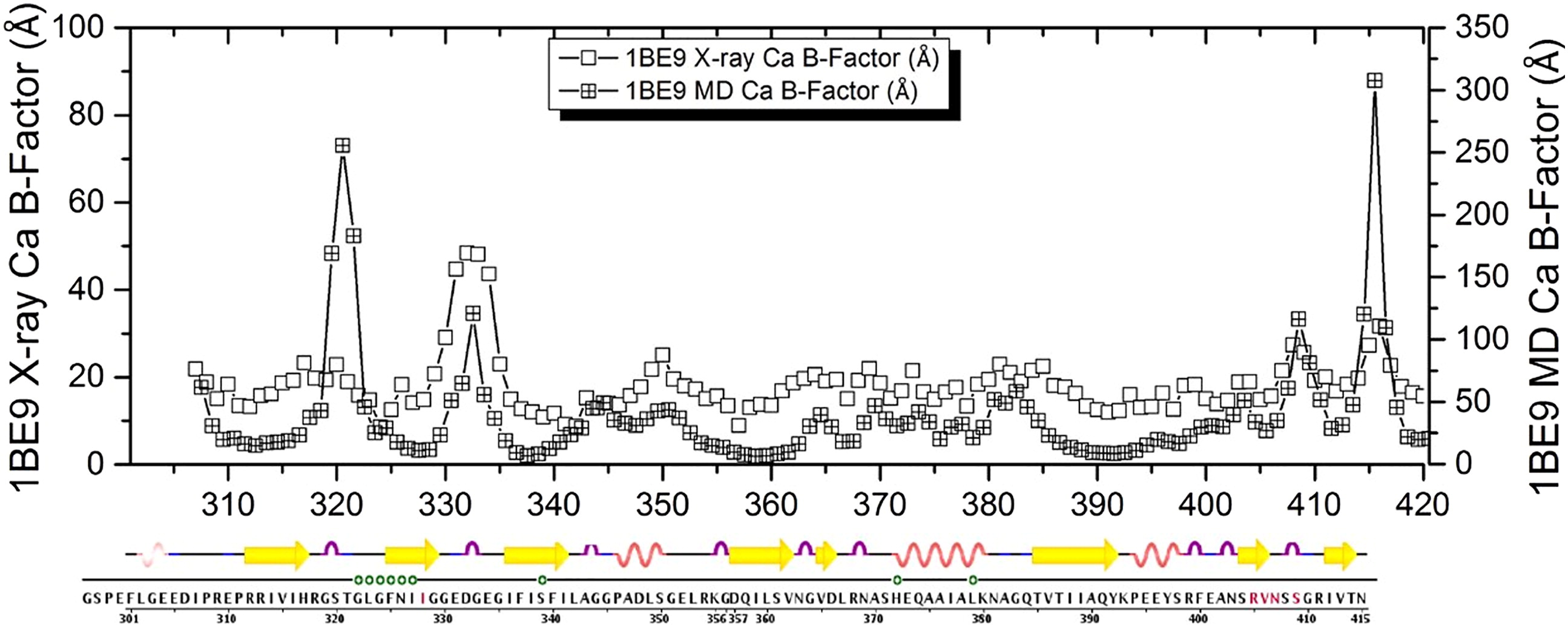 Spectral analysis of Molecular Dynamics computer simulations on the PDZ protein: MD: Sectors.
Spectral analysis of Molecular Dynamics computer simulations on the PDZ protein: MD: Sectors.
Lakhani B, Thayer KM, Beveridge DL.
Journal of Biomolecular Structure and Dynamics (2019). DOI: 10.1080/07391102.2019.1588169
The idea of protein “sectors” posits that sparse subsets of amino acid residues form cooperative networks that are key elements of protein stability, ligand binding, and allosterism. To date, protein sectors have been calculated by the statistical coupling analysis (SCA) method of Ranganathan and co-workers via the spectral analysis of conservation-weighted evolutionary covariance matrices obtained from a multiple sequence alignments of homologous families of proteins. SCA sectors, a knowledge-based protocol, have been indentified with functional properties and allosterism for a number of systems. -
Structural Determination of the Conformation of Glutathione Peroxidase-4 (GPX4) Through Markov State Modeling
Chung D, Thayer K, Beverdige D, Langley D.
The FASEB Journal, 33, 642 (2019). -
Dynamic Allosteric Biomolecular Design Using Artificial Intelligence
Thayer K, Beveridge D, Langley D.
The FASEB Journal, 33, 779 (2019). -
 Toward a Universal Structural and Energetic Model for Prokaryotic Promoters
Toward a Universal Structural and Energetic Model for Prokaryotic Promoters
Mishra A, Siwach, P, Mishra P, Jayaram B, Bansal M, Olson WK, Thayer KM, Beveridge, DL.
Biophys J (2018). DOI: 10.1016/j.bpj.2018.08.002.
With almost no consensus promoter sequence in prokaryotes, recruitment of RNA polymerase (RNAP) to precise transcriptional start sites (TSSs) has remained an unsolved puzzle. Uncovering the underlying mechanism is critical for understanding the principle of gene regulation. We attempted to search the hidden code in ∼16,500 promoters of 12 prokaryotes representing two kingdoms in their structure and energetics. -
 p53 tumor suppressor proteins R175H, G245S, R249S, and R282W: structural prediction and analysis of full length proteins
p53 tumor suppressor proteins R175H, G245S, R249S, and R282W: structural prediction and analysis of full length proteins
Thayer KM, Je L*.
MOJ Proteomics and Bioinformatics, 7, 92-100 (2018). DOI: 10.15406/mojpb.2018.07.00217 The tumor suppressor protein p53, the “Guardian of the Genome”1, prevents the development of cancerous tumors by removing or repairing cells with damaged genetic material. Approximately 50% or more of all human cancers are attributable to mutations in this protein.2,3 The p53 protein directs either of two major pathways through signaling for the DNA repair system, or, when the extent of damage is beyond repair, initiating programmed cell death, also known as apoptosis. -
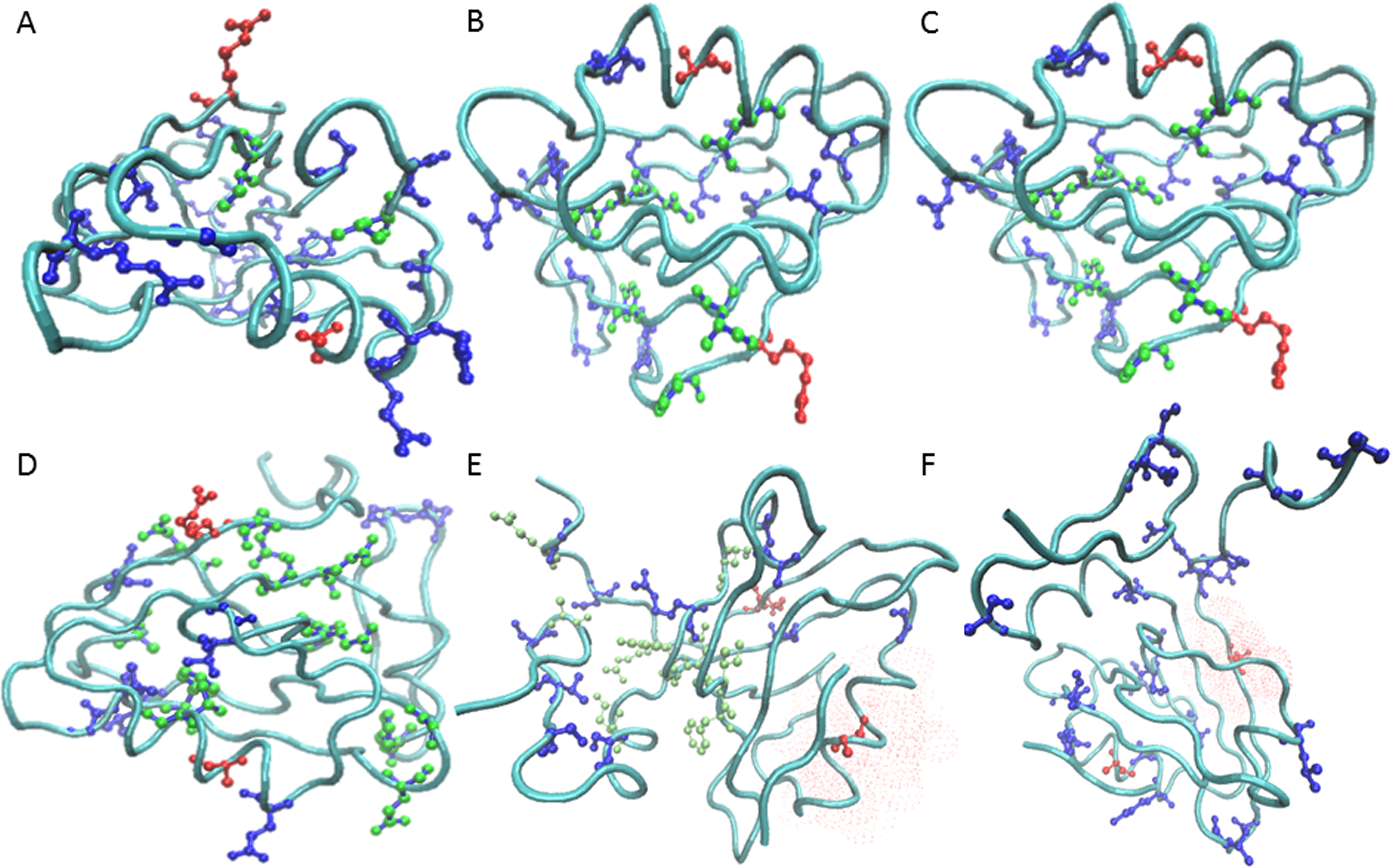 Dependence of prevalence of contiguous pathways in proteins on structural complexity.
Dependence of prevalence of contiguous pathways in proteins on structural complexity.
Thayer KM, Galganov JC*, Stein AJ*.
PLoS ONE, 12, e0188616 (2017). DOI 10.1371/journal.pone.0188616
Allostery is a regulatory mechanism in proteins where an effector molecule binds distal from an active site to modulate its activity. Allosteric signaling may occur via a continuous path of residues linking the active and allosteric sites, which has been suggested by large conformational changes evident in crystal structures. An alternate possibility is that the signal occurs in the realm of ensemble dynamics via an energy landscape change. While the latter was first proposed on theoretical grounds, increasing evidence suggests that such a control mechanism is plausible. -
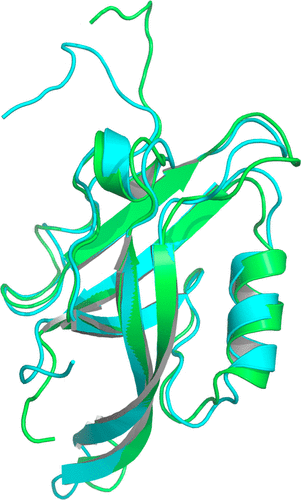 A Molecular Dynamics-Markov State Model of Protein Ligand Binding and Allostery in CRIB-PDZ: Conformational Selection and Induced Fit.
A Molecular Dynamics-Markov State Model of Protein Ligand Binding and Allostery in CRIB-PDZ: Conformational Selection and Induced Fit.
Thayer KM, Lakhani B, and Beveridge DL.
The Journal of Physical Chemistry B.,121, 5509-5514 (2017). DOI: 10.1021/acs.jpcb.7b02083
Conformational selection and induced fit are well-known contributors to ligand binding and allosteric effects in proteins. Molecular dynamics (MD) simulations now enable the theoretical study of protein–ligand binding in terms of ensembles of interconverting microstates and the population shifts characteristic of “dynamical allostery.” Here we investigate protein–ligand binding and allostery based on a Markov state model (MSM) with states and rates obtained from all-atom MD simulations. -
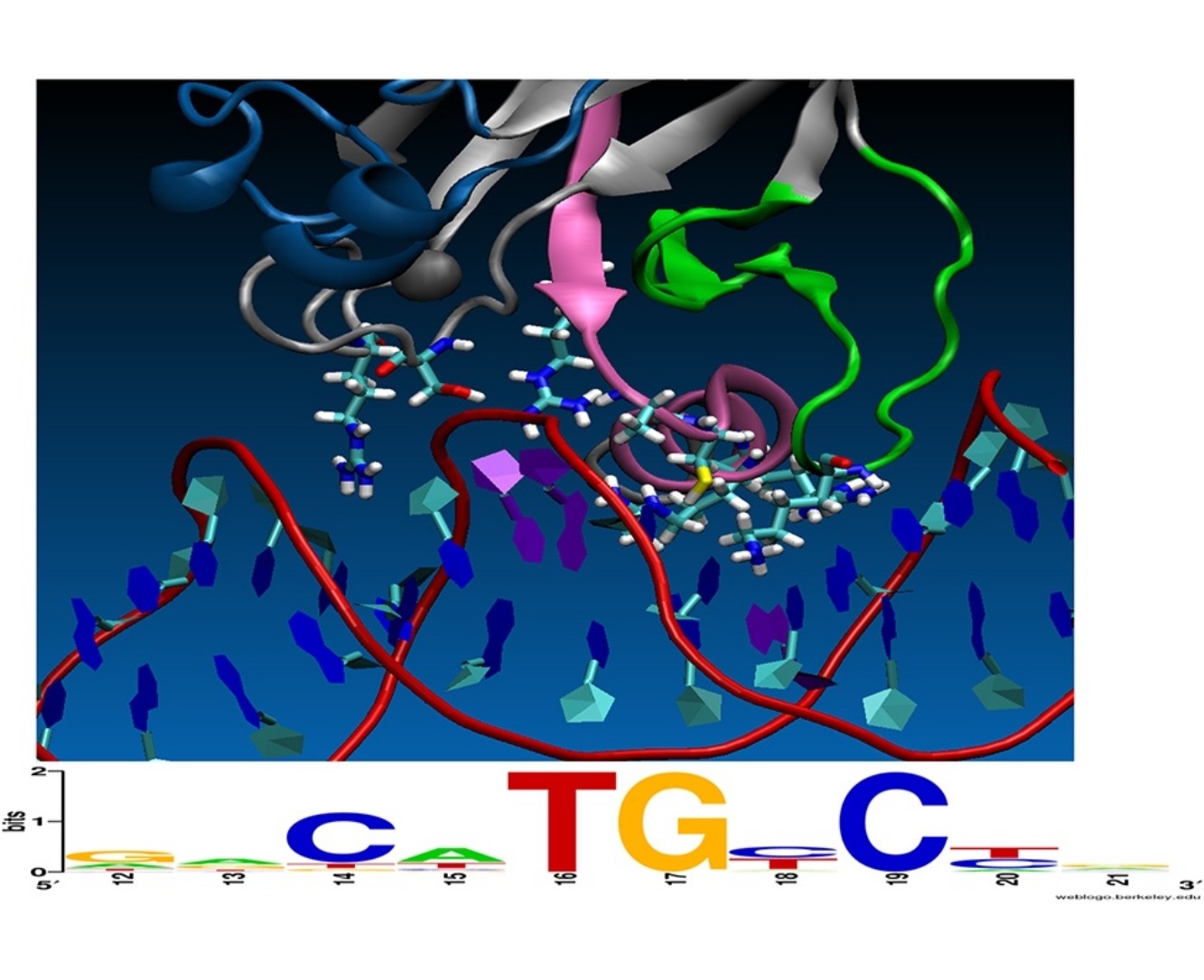 Chemical Principles Additive Model Aligns Low Consensus DNA Targets of p53 Tumor Suppressor Protein.
Chemical Principles Additive Model Aligns Low Consensus DNA Targets of p53 Tumor Suppressor Protein.
Thayer KM and Han IM*.
Computational Biology and Chemistry, 68, 186-193 (2017). DOI 10.1016/j.compbiolchem.2017.03.003
Computational prediction of the interaction between protein transcription factors and their cognate DNA binding sites in genomic promoters constitutes a formidable challenge in two situations: when the number of discriminatory interactions is small compared to the length of the binding site, and when DNA binding sites possess a poorly conserved consensus binding motif. The transcription factor p53 tumor suppressor protein and its target DNA exhibit both of these issues. -
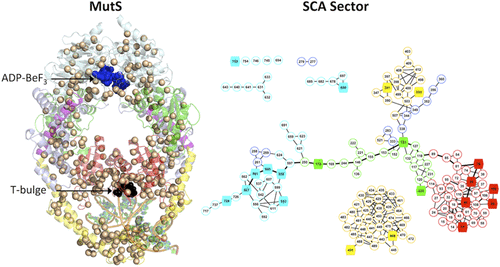 Evolutionary Covariance Combined with Molecular Dynamics Predicts a Framework for Allostery in the MutS DNA Mismatch Repair Protein.
Evolutionary Covariance Combined with Molecular Dynamics Predicts a Framework for Allostery in the MutS DNA Mismatch Repair Protein.
Lakhani B, Thayer KM, Hingorani MM, Beveridge DL.
The Journal of Physical Chemistry B., 191, 2049-2061 (2017). Mismatch repair (MMR) is an essential, evolutionarily conserved pathway that maintains genome stability by correcting base-pairing errors in DNA. Here we examine the sequence and structure of MutS MMR protein to decipher the amino acid framework underlying its two key activities—recognizing mismatches in DNA and using ATP to initiate repair. Statistical coupling analysis (SCA) identified a network (sector) of coevolved amino acids in the MutS protein family. -
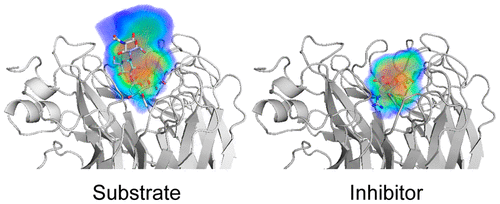 Molecular Basis for Differential Patterns of Drug Resistance in Influenza N1 and N2 Neuraminidase.
Molecular Basis for Differential Patterns of Drug Resistance in Influenza N1 and N2 Neuraminidase.
Prachanronarong KL, Ozen A, Thayer KM, Yilmaz LS, Zeldovich KB, Bolon DN, Kowalik TF,
Jensen JD, Finberg RW, Wang JP, Kurt-Yilmaz N, Schiffer CA.
Journal of Chemistry and Theoretical Chemistry, 12, 6098-6108 (2016).
Neuraminidase (NA) inhibitors are used for the prevention and treatment of influenza A virus infections. Two subtypes of NA, N1 and N2, predominate in viruses that infect humans, but differential patterns of drug resistance have emerged in each subtype despite highly homologous active sites. To understand the molecular basis for the selection of these drug resistance mutations, structural and dynamic analyses on complexes of N1 and N2 NA with substrates and inhibitors were performed. -
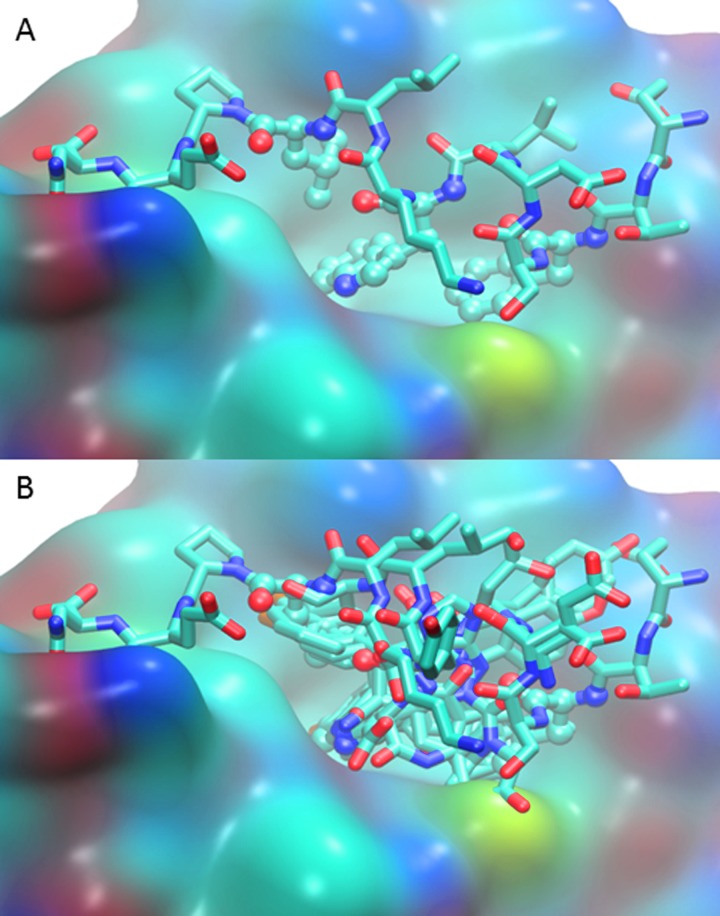 Energetic Landscape of MDM2-p53 Interactions by Computational Mutagenesis of the MDM2-p53 Interaction Interface.
Energetic Landscape of MDM2-p53 Interactions by Computational Mutagenesis of the MDM2-p53 Interaction Interface.
Thayer KM and Beyer GA.*
PLoS ONE, 11, e0147806 (2016).
The ubiquitin ligase MDM2, a principle regulator of the tumor suppressor p53, plays an integral role in regulating cellular levels of p53 and thus a prominent role in current cancer research. Computational analysis used MUMBO to rotamerize the MDM2-p53 crystal structure 1YCR to obtain an exhaustive search of point mutations, resulting in the calculation of the ΔΔG comprehensive energy landscape for the p53-bound regulator. -
Computational Construction of a p53 Tetrameric Structure.
Thayer KM and Carpenter C.*
MOJ Proteomics and Bioinformatics, 3, 71 (2016). -
p53 R175H Hydrophobic Patch and H-Bond Reorganization Observed by MD Simulation.
Thayer KM and Quinn, TR.*
Biopolymers, 105, 176-185 (2016).
Molecular dynamics simulations probe the origins of aberrant functionality of R175H p53, which normally prevent tumorigenesis. This hotspot mutation exhibits loss of its essential zinc cofactor, aggregation, and activation of gain of function promoters, characteristics contributing to the loss of normal p53 activity. This study provided molecular level insight into the reorganization of the hydrogen bonding network and the formation of a hydrophobic patch on the surface of the protein. -
Structural point mutations of p53 protein and their effects on the zinc coordination complex
Quinn T*, Thayer K.
The FASEB Journal 29, 712.23 (2015).
The p53 protein is an important transcription factor in the cell cycle. As such, mutations of this protein are found in at least 50% of all cancers, most of which are in the DNA-binding domain containing residues 102-292. This research is focusing on the molecular dynamics of p53 with point mutations that affect the structural stability of the protein, namely, R175H, R282W, G245S, and R249S. -
Molecular Dynamics of DNA Mechanical Contributions of Various Promoter Sequences in p53 Complexes Motivated by a Novel Additive Binding Energy Model
Han IS* and Thayer KM.
The FASEB Journal 29, 712.22(2015).
The tumor suppressor protein p53 binds genomic DNA as a cell cycle regulator that ensures DNA gets repaired before replication. When excessive damage renders cells non-salvageable, the protein initiates apoptosis; however, disruption of this process may lead to tumor growth. An additive binding-energy model was developed as a means to categorize known DNA binding sequences based on their hydrogen bonding patterns and DNA bending mechanics. -
Characterization of the Readout Mechanism of the L1 Loop’s Role in the p53 Tumor Suppressor Protein Binding Event via Molecular Dynamics Simulations
Slaw B* and Thayer K.
The FASEB Journal 29, 712.24(2015).
A primary target in cancer research, the p53 tumor suppressor protein, is a transcription activator playing an integral role in the regulation of the cell cycle. Through binding to DNA, the protein has the ability to “turn on” other repair proteins to mend damaged DNA, inhibit cellular division, and initiate apoptosis in cells that are damaged beyond repair. -
Total Mutagenesis and the Effects on the MDM2-p53 Interaction
Beyer G* and Thayer K.
The FASEB Journal 29, 712.19(2015).
While the available literature explores alanine-scanning mutagenesis of the six necessary amino acids of the p53 binding subdomain in order to obtain precise ΔG values for the wild-type amino acids in the p53-MDM2 binding domain, the effects of non-alanine point mutations have not been explored. Many mutations of MDM2 that could affect the binding to p53 have largely gone unstudied, but several SNPs in MDM2 have been directly linked to tumor formation. -
Structural bioinformatics and molecular dynamics of p53 tumor suppressor protein
Thayer K, Slaw B*, Han I*, Quinn T*.
The FASEB Journal 29, 712.12 (2015).
In carrying out its tumor suppressor function, p53 binds to cognate DNA regulatory sites throughout the genome. While many of the binding sites contain a recognizable consensus, other binding sites exist which lack this, making this an interesting system for understanding the potential role of indirect readout in the binding event. In order to study this, we are constructing additive energy models based on hydrogen bond contacts and the presence of the TG DNA base pair step. -
Diversity of T Cell Epitopes in Plasmodium falciparum Circumsporozoite Protein Likely Due to Protein-Protein Interactions.
Aragam NA**, Thayer KM**, Nge N, Hoffman I, Martinson F, Kamwendo D, Lin FC, Sutherland
C, Bailey JA, Juliano JJ. (**co-first authors)
PLoS ONE, 8,62427 (2013).
**equal contributions as first authors -
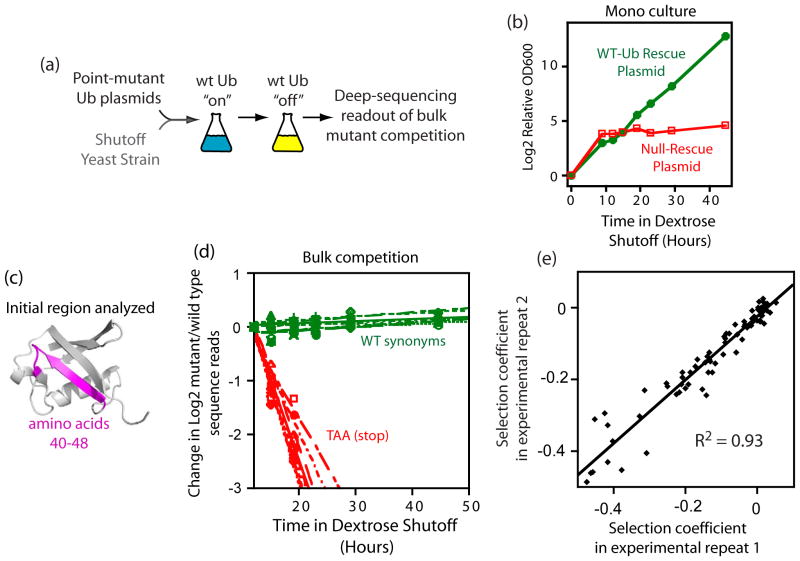 Analyses of the effects of all ubiquitin point mutants on yeast growth rate.
Analyses of the effects of all ubiquitin point mutants on yeast growth rate.
Roscoe BP, Thayer KM, Zeldovich KB, Fushman D, Bolon DN.
J Mol. Biol., 425, 1363 (2013).
The amino acid sequence of a protein governs its function. We used bulk competition and focused deep sequencing to investigate the effects of all ubiquitin point mutants on yeast growth rate. Many aspects of ubiquitin function have been carefully studied, which enabled interpretation of our growth analyses in light of a rich structural, biophysical and biochemical knowledge base. In one highly sensitive cluster on the surface of ubiquitin, almost every amino acid substitution caused growth defects. In contrast, the opposite face tolerated virtually all possible substitutions. -
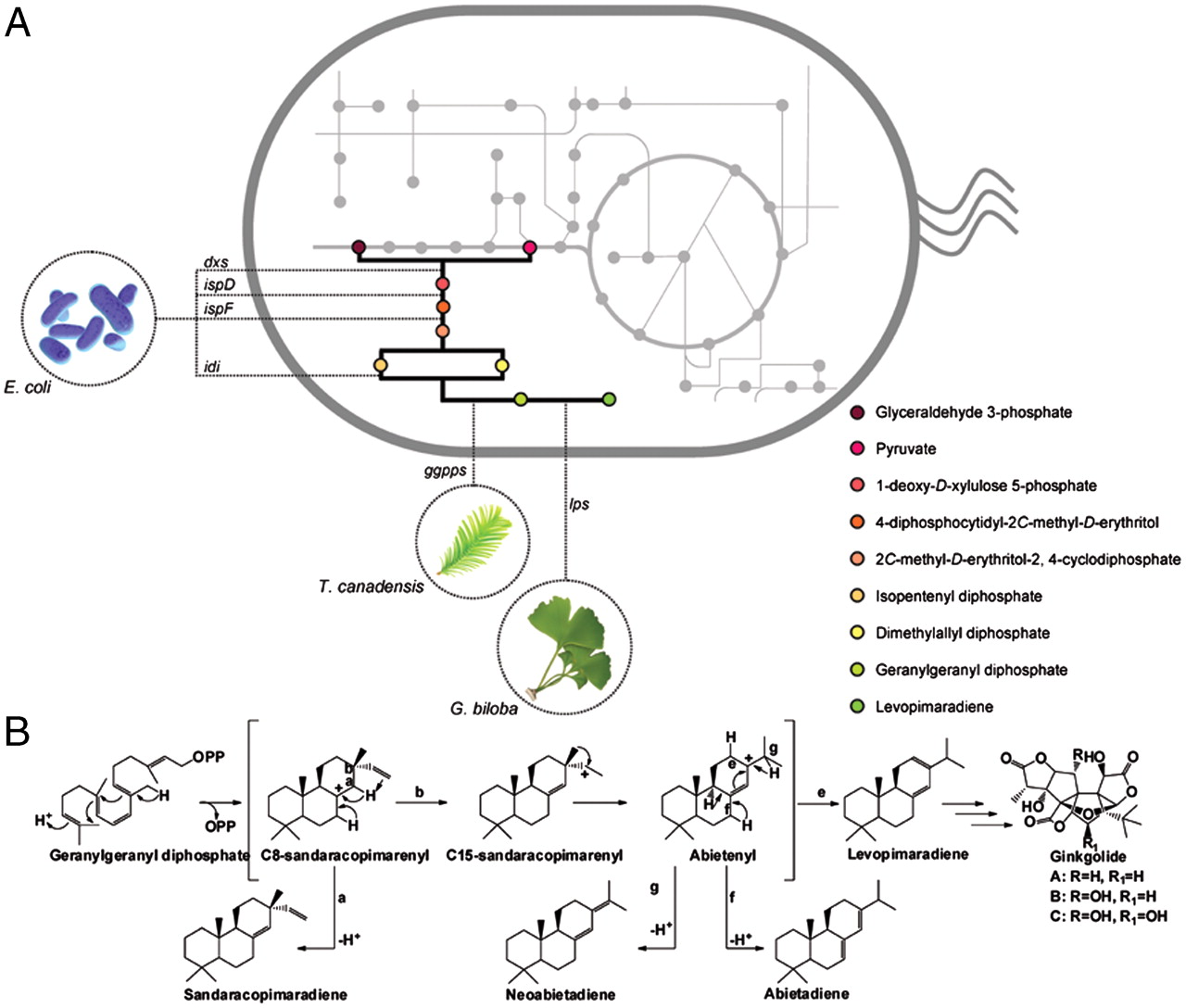 Combining metabolic and protein engineering of a terpenoid biosynthetic pathway for overproduction and selectivity control.
Combining metabolic and protein engineering of a terpenoid biosynthetic pathway for overproduction and selectivity control.
Leonard E, Ajikumar PK, Thayer K, Xiao WH, Mo JD, Ditor B, Stephanopoulos G, Prather KL.
Proceedings of the National Academy of Sciences USA, 107, 13654-9(2010).
A common strategy of metabolic engineering is to increase the endogenous supply of precursor metabolites to improve pathway productivity. The ability to further enhance heterologous production of a desired compound may be limited by the inherent capacity of the imported pathway to accommodate high precursor supply. Here, we present engineered diterpenoid biosynthesis as a case where insufficient downstream pathway capacity limits high-level levopimaradiene production in Escherichia coli. -
Molecular Basis for Drug Resistance in HIV-1 Protease.
Ali A, Bandaranayake RM, Cai Y, King NM, Kolli, M, Mittal S, Murzycki JF, Nalam MN, Nalivaika
EA, Ozen A, Prabu-Jeyabalan MM, Thayer K, Schiffer CA.
Viruses, 2, 2509 (2010).
HIV-1 protease is one of the major antiviral targets in the treatment of patients infected with HIV-1. The nine FDA approved HIV-1 protease inhibitors were developed with extensive use of structure-based drug design, thus the atomic details of how the inhibitors bind are well characterized. From this structural understanding the molecular basis for drug resistance in HIV-1 protease can be elucidated. -
Molecular Dynamics Simulations of the 136 Unique Tetranucleotide Sequences of DNA
Oligonucleotides. II: Sequence context effects on the dynamical structures of the 10 unique
dinucleotide steps.
Dixit SB, Beveridge DL, Case DA, Cheatham III TE, Giudice E, Lankas F, Lavery R, Maddocks JH,
Osman R, Sklenar H, Thayer KM, Varnai P.
Biophysical Journal, 89, 3271 (2005). -
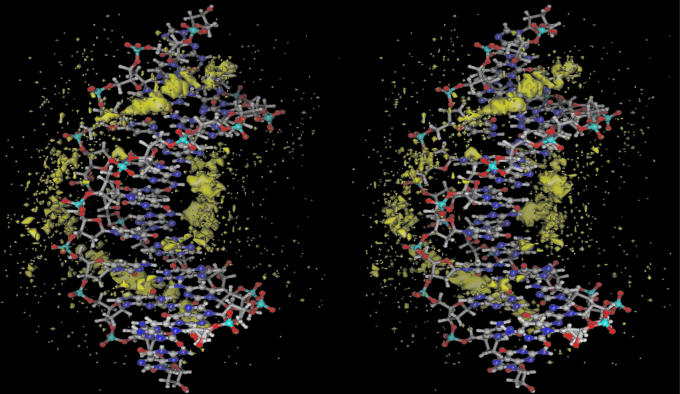 Ion motions in molecular dynamics simulations on DNA.
Ion motions in molecular dynamics simulations on DNA.
Ponomarev SY, Thayer KM, Beveridge DL.
Proceedings of the National Academy of Sciences USA, 101, 14771-5 (2004).
Counterions play a significant role in DNA structure and function, and molecular dynamics (MD) simulations offer the prospect of detailed description of the dynamical structure of ions at the molecular level. However, the motions of mobile counterions are notably slow to converge in MD on DNA. Obtaining accurate and reliable MD simulations requires knowing just how much sampling is required for convergence of each of the properties of interest. -
Molecular dynamics simulations of DNA curvature and flexibility: Helix phasing and premelting.
Beveridge DL, Dixit SB, Barreiro G, Thayer KM.
Biopolymers, 73, 380-403 (2004).
Recent studies of DNA axis curvature and flexibility based on molecular dynamics (MD) simulations on DNA are reviewed. The MD simulations are on DNA sequences up to 25 base pairs in length, including explicit consideration of counterions and waters in the computational model. MD studies are described for ApA steps, A-tracts, for sequences of A-tracts with helix phasing. In MD modeling, ApA steps and A-tracts in aqueous solution are essentially straight, relatively rigid, and exhibit the characteristic features associated with the B'-form of DNA. -
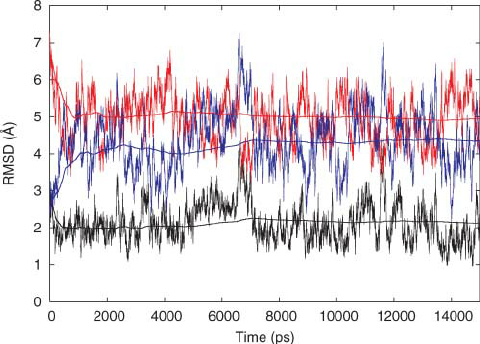 Molecular dynamics simulations of the 136 unique Tetranucleotide sequences of DNA oligonucleotides. I: Research design and results on d(CpG) steps.
Molecular dynamics simulations of the 136 unique Tetranucleotide sequences of DNA oligonucleotides. I: Research design and results on d(CpG) steps.
Beveridge DL, Barreiro G, Byun KS, Case DA, Cheatham III TE, Dixit SB, Giudice E, Lankas F,
Lavery R, Maddocks JH, Osman R, Seibert E, Sklenar H, Stoll G, Thayer KM, Varnai P, Young
MA.
Biophysical Journal, 87, 3799-813 (2004).
We describe herein a computationally intensive project aimed at carrying out molecular dynamics (MD) simulations including water and counterions on B-DNA oligomers containing all 136 unique tetranucleotide base sequences. This initiative was undertaken by an international collaborative effort involving nine research groups, the "Ascona B-DNA Consortium" (ABC). -
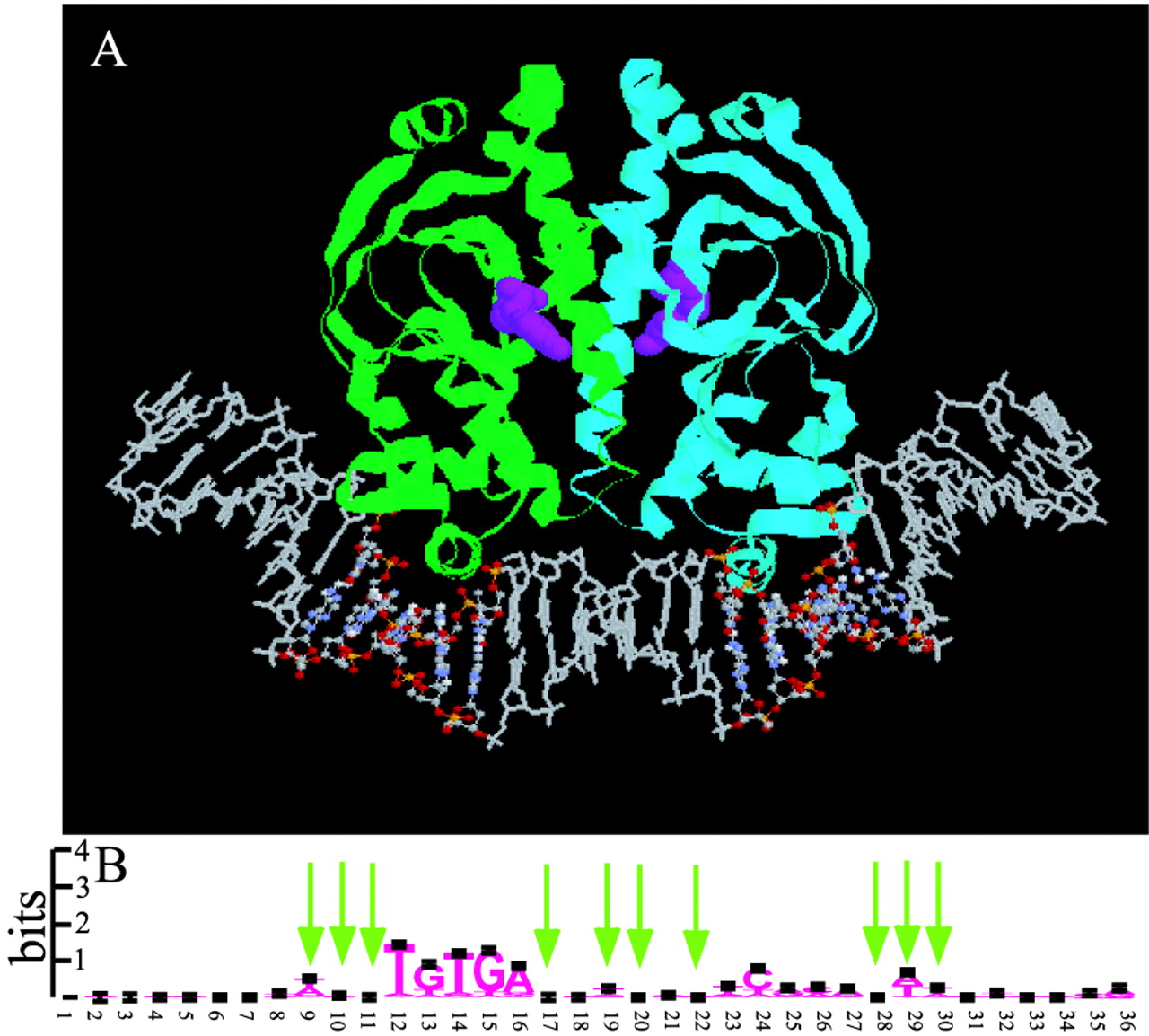 Hidden Markov models from molecular dynamics simulations on DNA
Hidden Markov models from molecular dynamics simulations on DNA
Thayer KM and Beveridge DL.
Proceedings of the National Academy of Sciences USA, 99, 8642-8647 (2002).
An enhanced bioinformatics tool incorporating the participation of molecular structure as well as sequence in protein DNA recognition is proposed and tested. Boltzmann probability models of sequence-dependent DNA structure from all-atom molecular dynamics simulations were obtained and incorporated into hidden Markov models (HMMs) that can recognize molecular structural signals as well as sequence in protein–DNA binding sites on a genome. The binding of catabolite activator protein (CAP) to cognate DNA sequences was used as a prototype case for implementation and testing of the method.